



Angioedema hereditario, enfermedad rara, genética e incurable, pero tratable
Poco conocimiento de esta enfermedad ocasiona mala calidad de vida en el paciente. Eventos de hinchazón en membranas de mucosas respiratorias pueden ser mortales
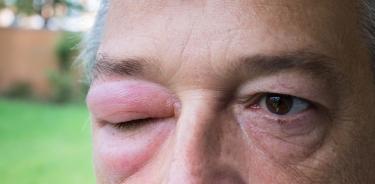
Persona con sintomatología de angioedema hereditario
En el mundo se tienen identificadas alrededor de seis mil enfermedades denominadas raras, o de baja prevalencia –aunque en nuestro país, el sistema de salud apenas reconoce 20-, llamadas así debido al bajo número de personas que se ven afectadas por esta condición.
En México, datos de la Secretaría de Salud señalan que alrededor de ocho millones de personas se ven afectadas por alguna de estas enfermedades también conocidas como “huérfanas”, y consideradas así, cuando no hay más de cinco mil personas con alguna enfermedad rara, por cada 100 mil habitantes.
Entre este tipo de padecimientos, se encuentra el angioedema hereditario (AEH), enfermedad que tiene una baja prevalencia estimada a nivel mundial en el rango de uno por cada 50 mil habitantes o llega a ser incluso de uno por cada 100 mil habitantes y puede afectar por igual a hombres o mujeres.
El problema con el angioedema hereditario -enfermedad origen genético, por un bajo nivel o mal funcionamiento de una proteína llamada inhibidor de C1, que afecta prácticamente a todos los vasos sanguíneos-, es que cómo ocurre con muchas de estas afecciones raras, es que se confunde con otro tipo de padecimientos y en consecuencia se dan diagnósticos erróneos, postergando así, la posibilidad de que el paciente con AEH pueda iniciar en etapas tempranas un tratamiento adecuado.
Al respecto, la doctora Estefanía Torres, líder del Área Terapéutica de Enfermedades Genéticas en Takeda México, explicó que esta enfermedad, es una condición la cual se caracteriza por episodios recurrentes y dolorosos de hinchazón que afectan la piel y las membranas mucosas del sistema respiratorio y gastrointestinal.
Algunos de los síntomas que se han identificado son: obstrucción de vías respiratorias, que provoca también inflamación de garganta, episodios repetitivos de cólicos abdominales sin causa aparente, hinchazón en manos, garganta, brazos, piernas, labios, ojos, lengua, así como hinchazón intestinal que puede ser grave que puede provocar cólicos abdominales, vómito, diarrea.
La hinchazón puede aparecer de manera impredecible, ocasionando un importante impacto en la calidad de vida de estos pacientes que lo padecen, causando malestar y, en casos graves, incluso provocar la muerte, cuando es en el sistema respiratorio, porque, se presenta en mucosas de las vías respiratorias altas y tracto gastrointestinal.
Esto ocurre, porque la deficiencia en la proteína inhibidor de C1 esterasa, el organismo es incapaz de poder regula algún proceso inflamatorio.
El paciente que no es diagnosticado de manera adecuada a tiempo, dijo, llega a tener una sobrevida de 22 años en promedio y un impacto significativo en su calidad de vida antes de obtener finalmente un diagnóstico correcto y un tratamiento adecuado.
Refirió que en ocasiones, algunos pacientes son sometidos a cirugías innecesarias, como la apendicectomía, debido a la falta de conciencia sobre la enfermedad, y suele confundirse con apendicitis.
Es importante mencionar que, se estima que en el país hay más de 2,500 personas con AEH, aunque apenas poco más de 300 están plenamente identificadas y diagnosticada, y sin embargo, menos del 20% reciben el tratamiento adecuado.
Respecto a los tratamientos disponibles, se cuenta con cuatro medicamentos específicos para el angioedema hereditario, dos son para eventos agudos, de emergencia, ante edemas en la garganta, uno de aplicación intravenoso y el otro subcutáneo, y otros dos medicamentos de profilaxis, para evitar que los pacientes presenten alguna crisis, sobre todo en quienes tienen eventos recurrentes.